The research team “Genetic causes of movement disorders” focuses on the identification and evaluation of mutations and genetic risk factors in different movement disorders with a primary focus on dystonia but also on Parkinson disease, restless legs syndrome, and ataxias. Moreover, we are interested in the causes of neurodevelopmental disorders.
Major achievements of this group include the identification of genetic risk factors for musician’s dystonia, and the identification of novel disease genes by exome and genome sequencing, including TUBB4A, RAB12, GNB1, VPS13D, and TAOK1. Next generation sequencing techniques are currently also used to reveal the underlying genetic causes in other families as well as sporadic patients. In addition, several hundred Parkinson´s disease and dystonia patients from cohort studies such as EPIPARK and registers such as DYSTRACT are being screened for mutations in known disease genes by Gene Panel analyses leading to the identification of mutation carriers who are being deeply clinically characterized.
Another research focus lies on the characterization of the dystonia-causing gene THAP1 and its encoded protein. We identified several disease-causing mutations in THAP1 and performed functional studies of the mutants. Further, we demonstrated that THAP1 functions as a transcription factor that regulates the expression of the DYT-TOR1A dystonia-causing gene TorsinA in-vitro as well as its own expression. Recently, we generated iPSC-derived cortical neurons of ten affected and unaffected THAP1 mutation carriers to study transcriptional dysregulation in disease-related cells.
Projects are mainly funded by the German Research Foundation (DFG), the Federal Ministry of Education and Sciences (BMBF), and the Damp Foundation.
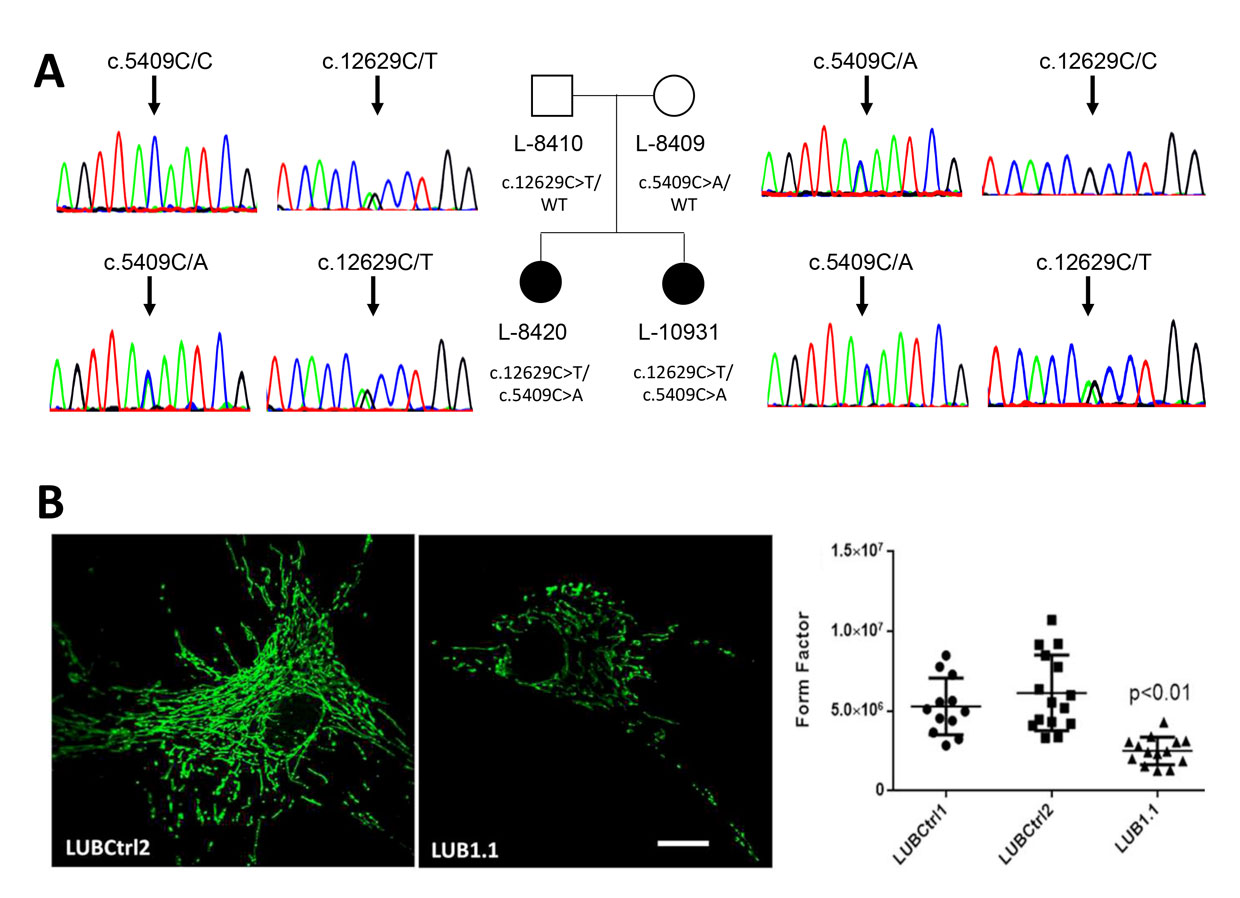
Figure: Identification and functional characterization of compound heterozygous pathogenic variants in VPS13D as cause of a spastic ataxia in two siblings. (A) The two affected sisters in the family are indicated by black filled symbols. They both carry two mutations in the VPS13D gene that are illustrated by the respective electropherograms from Sanger sequencing. Their healthy parents carry each only one of the mutations as shown. (B) The mitochondrial network was stained by immunocytochemistry with an anti-GRP75 antibody in cultures of patient-derived fibroblasts. The mitochrondria in the patient (LUB1.1) appear smaller, rounder, and less connected when compared to a mutation-negative control (LUBCtrl2). The mitochondrial network was quantified in at least twelve cells of two different control lines (LUBCtrl1 and 2) and the patient by form factor analyses. This revealed a significantly altered mitochrondrial network in the patient. (published in Seong et al. Ann Neurol 2018, PMID: 29604224)